Samenvatting
Multipele epifysaire dysplasie is een zeldzame autosomaal dominant overervende afwijking van de epifysaire schijven die tot ernstige gewrichtsdestructies leidt. Een grote Nederlandse familie met multipele epifysaire dysplasie wordt beschreven; de belangrijkste klinische verschijnselen manifesteerden zich aan de onderste extremiteiten.
In verband met verschillen in therapeutische benadering moet multipele epifysaire dysplasie onderscheiden worden van andere ziekten met gewrichtsafwijkingen.
artikel
Inleiding
Erfelijke afwijkingen van het steun- en bewegingsapparaat die zich op latere leeftijd manifesteren, zijn relatief zeldzaam. Eén ervan is multipele epifysaire dysplasie (MED), een ossificatiestoornis van de epifyse van pijpbeenderen die leidt tot een ernstige deformatie van het betrokken gewricht. Het ziektebeeld is voor het eerst beschreven in 1947 als ‘dysplasia epiphysealis multiplex’.1 Wanneer ook de wervels aangedaan zijn, wordt gesproken van spondylo-epifysaire dysplasie.
MED is een autosomaal dominant overervende aandoening met een hoge penetrantie en variabele expressie.2 De aandoening veroorzaakt vanaf de vroege jeugd destructies van de grote gewrichten van de bovenste en de onderste extremiteiten. Wij onderzochten een grote familie met deze aandoening, met overwegend klachten van de onderste ledematen, uit onze praktijk (St. Joseph Ziekenhuis te Veldhoven). Slechts éénmaal eerder werd in de literatuur een dergelijke familie beschreven.
PatiËnten en methoden
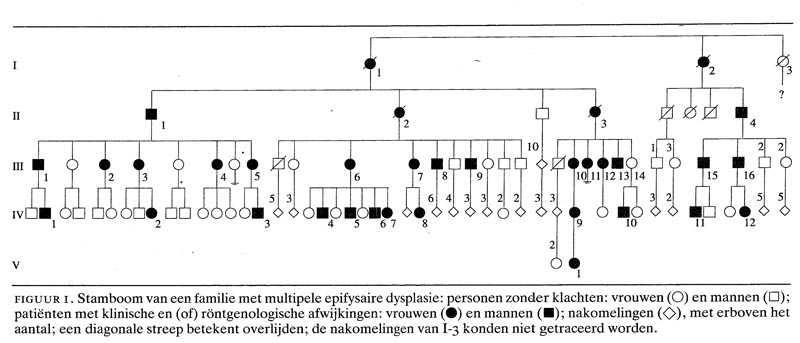
De stamboom van de familie staat in figuur 1.
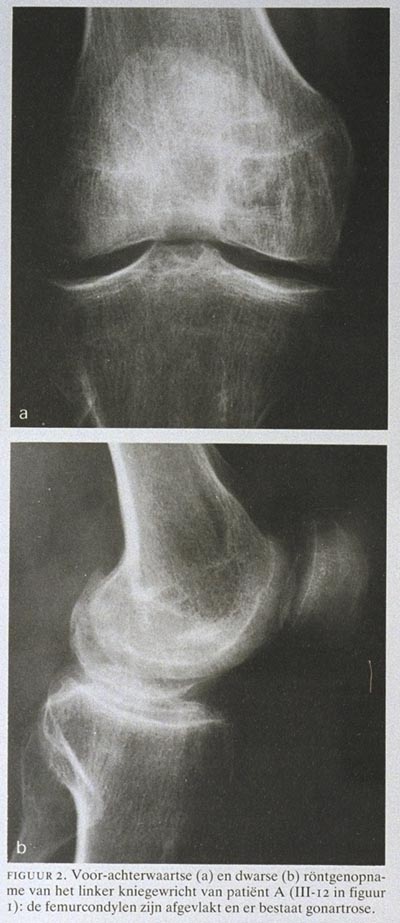
Patiënt A was een 54-jarige vrouw (III-12 in figuur 1), sinds vele jaren onder behandeling wegens elleboog- en knieklachten. Meerdere malen werd zij geopereerd, waarbij artroscopieën en gewrichtsnettoyages werden uitgevoerd. Haar moeder, inmiddels overleden, liep altijd moeilijk en had knieorthesen. Haar 27-jarige dochter had geen klachten. Bij onderzoek werd een moeilijk lopende vrouw gezien met genua vara en beperkte functie van elleboog- en kniegewrichten. Röntgenopnamen van het kniegewricht (figuur 2) bevestigden de diagnose MED: de femurcondylen zijn afgevlakt en er is sprake van gonartrose.
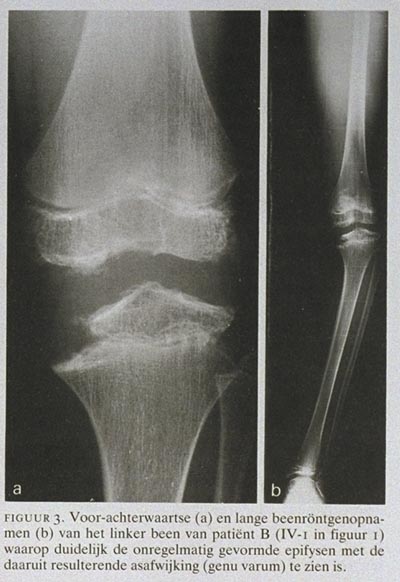
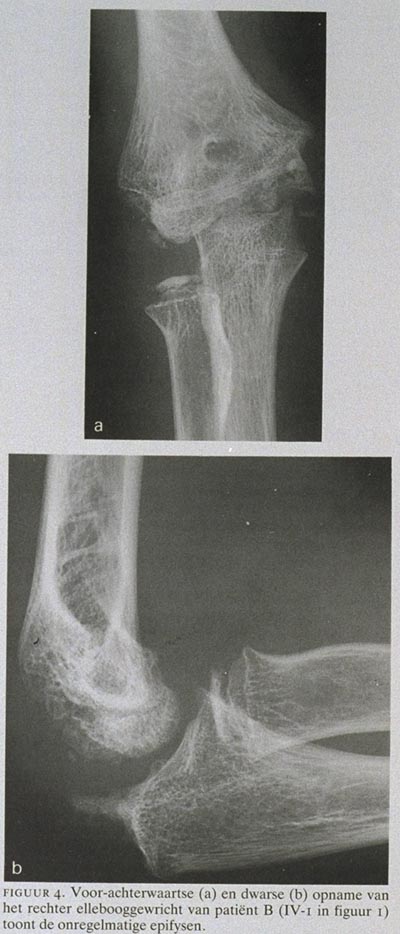
Patiënt B, een 15-jarige jongen (IV-1 in figuur 1), had vanaf zijn 6e jaar de klachten dat hij niet goed kon rennen en een stijf gevoel in de gewrichten had. Hij was langdurig met fysiotherapie behandeld. Vanaf het 12e levensjaar namen de klachten toe. Patiënt had pijn in de knieën zonder slotklachten. Bij onderzoek werden duidelijk genua vara gezien. Op röntgenfoto's van de knieën (figuur 3) zijn de onregelmatige, kleine epifysen met verbreding van de metafysen te zien; op lange beenopnamen is de asafwijking te zien. Op röntgenfoto's van de ellebogen (figuur 4), eveneens gemaakt op 12-jarige leeftijd, zijn de onregelmatig gevormde gewrichtsoppervlakken en artrose te zien. Van de ellebooggewrichten had patiënt evenwel geen last. Zijn vader had al vele jaren knieklachten, zijn broer had geen last van de gewrichten.
In verband met het hereditaire karakter van MED werd naar aanleiding van de bevindingen bij deze 2 patiënten uitgebreid familie-onderzoek verricht. Er kon een zeer uitgebreide stamboom opgesteld worden (zie figuur 1).
Resultaten
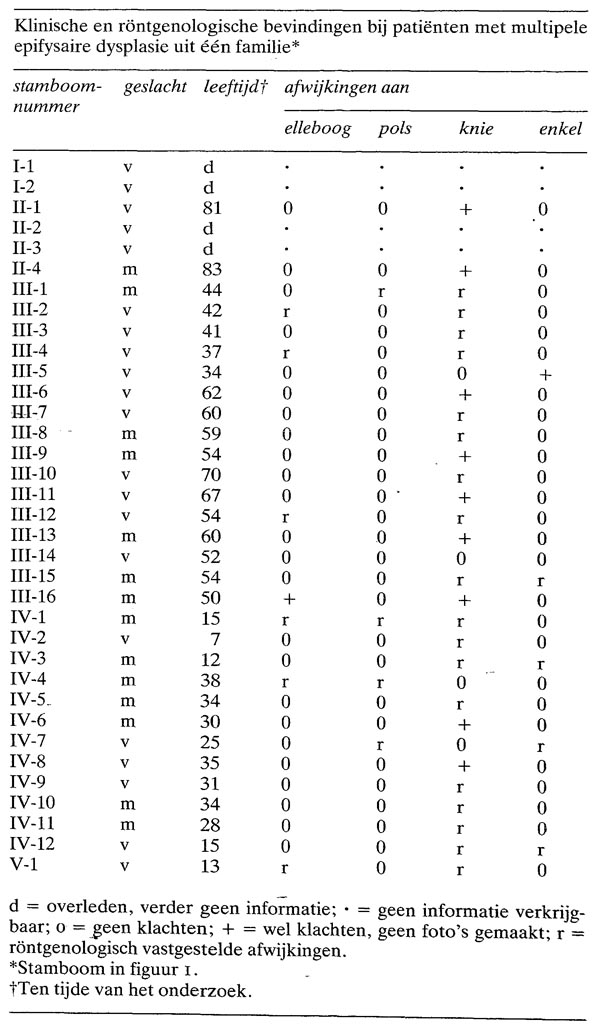
De familie omvatte meer dan 150 leden, van wie er 34 MED hadden op grond van de klinische en (of) röntgenologische afwijkingen (zie figuur 1). De nakomelingen van 1-3 konden niet getraceerd worden. Afwijkingen aan de knieën kwamen bij 27 patiënten voor, aan de ellebogen bij 7, aan de handen bij 4 en aan de enkels bij 5. Een samenvatting van de klinische en de röntgenologische gegevens is weergegeven in de tabel. Daarin is ook familielid III-14 opgenomen, een vrouw zonder klachten met een zoon die wel klachten had.
Beschouwing
MED begint meestal in de vroege jeugd met loopstoornissen. Vooral heup-, knie-, enkel-, schouder- en polsgewrichten zijn aangedaan, in principe bilateraal en bij beide seksen even vaak. Later treden asafwijkingen op en kunnen corpora libera leiden tot slotverschijnselen. Op volwassen leeftijd wordt het klinische beeld rustiger en resteren de ontstane asafwijkingen en artrose.
Op röntgenfoto's zijn bij jonge kinderen meerdere ossificatiecentra in de groeischijf zichtbaar; de ossificatiecentra verschijnen laat en zijn vaak afgevlakt. De epifyse heeft een onregelmatig, gevlekt aspect. In de loop der jaren verbetert het röntgenologische beeld. De epifysaire uiteinden van de aangedane botten krijgen een regelmatige, normale botstructuur. Op volwassen leeftijd zijn voornamelijk de articulerende oppervlakken afwijkend. Deze zijn dan afgevlakt, irregulair en soms wigvormig. Later treedt vaak secundaire artrose op.3
De oorzaak van het ontstaan van MED is niet bekend. De primaire stoornis bevindt zich in de chondrocyt van de epifyse, waardoor vertraagde en onregelmatige ossificatie van de epifysaire uiteinden van de botten optreedt. De periferie van de epifyse wordt irregulair en de zo ontstane gewrichtsincongruentie leidt tot secundaire artrose.
Uit de stamboom (zie figuur 1) blijkt de autosomaal dominante overerving. De stamboom is van een zelden eerder beschreven omvang. De prevalentie van MED wordt in de literatuur geschat op 11-16 per miljoen;2 naar alle waarschijnlijkheid is dit een onderschatting vanwege het soms lichte beloop van de aandoening, waardoor bezoek aan een arts achterwege blijft. De penetrantie van MED is groot, doch niet volledig: een 52-jarige vrouw (III-14) was geheel zonder klachten, terwijl haar 34-jarige zoon evident artrotische veranderingen heeft, passend bij MED.
De variabele expressie van MED komt tot uiting in het type gewricht en in de mate waarin dit gewricht aangedaan is. In de door ons onderzochte familie werden voornamelijk knieklachten gezien en geen schouder- of heupafwijkingen. De andere grote familie die in de literatuur is beschreven (7 generaties, 163 leden en 53 patiënten) vertoonde vooral afwijkingen aan de onderste extremiteiten.4 Een mogelijke verklaring voor het slechts in bepaalde gewrichten vóórkomen van het ziektebeeld is genetische heterogeniteit of het bestaan van multipele allelen op één gen.5
De afwijkingen in de door ons beschreven familie waren relatief licht. Ernstige macroscopische afwijkingen, bijvoorbeeld indrukwekkende asafwijkingen of korte lichaamsbouw, kwamen in deze familie niet voor.
Differentiaaldiagnostisch moet MED onderscheiden worden van spondylo-epifysaire dysplasie, osteochondritis dissecans of, in geval van heupafwijkingen, van de ziekte van Perthes. Osteochondritis dissecans ontwikkelt zich in een verder normale epifyse, is pijnlijk en wordt ernstiger wanneer geen adequate behandeling gegeven wordt. De gewrichtsdestructies bij MED zijn veel ernstiger en meer verspreid over het gehele gewricht, de ziekte is altijd bilateraal en tast vaak meerdere gewrichten aan. De ziekte van Perthes is meestal eenzijdig gelokaliseerd en beperkt tot het caput femoris, terwijl bij MED ook het acetabulum afwijkingen vertoont.6
Het is differentiaaldiagnostisch van belang te letten op extraskeletale afwijkingen zoals myopie, doofheid, abnormale beharing en dysmorfie van het gelaat. Andere erfelijke aandoeningen met gewrichtsafwijkingen, zoals het trichorinofalangeaal syndroom, type I, moeten uitgesloten worden.
Wanneer MED leidt tot losse kraakbeenhaarden dient de behandeling te bestaan uit het verwijderen van deze haarden, gevolgd door continue passieve beweging zodat remodellering op kan treden. In een later stadium zijn vaak corrigerende osteotomieën en gewrichtsnettoyages nodig, of zelfs gewrichtsprothesen. Het is onjuist om als behandeling op jonge leeftijd gedeeltelijk losse kraakbeenhaarden te verwijderen zonder verdere behandeling of, in het geval van heupafwijkingen, de heupgewrichten in abductie-orthesen te immobiliseren.
Conclusie
MED is een betrekkelijk onbekend ziektebeeld dat zeer ernstige gewrichtsdestructies tot gevolg kan hebben. De behandeling dient aanvankelijk zoveel mogelijk conservatief te zijn en moet bestaan uit oefentherapie.
Preventieve adviezen, gericht op gewichtsreductie en verantwoorde beroepskeuze, zijn belangrijk. Verder is het van belang patiënten op de hoogte te stellen van de erfelijkheid van de ziekte. Wij adviseren een verwijzing naar een klinisch genetisch centrum.
Met dank aan mw.C.E.M.de Die-Smulders, klinisch geneticus, voor het kritisch reviseren van het manuscript.
Literatuur
Fairbank HAT. Dysplasia epiphysealis multiplex. Br J Surg1947; 34: 225-32.
Tachdjian MO. Pediatric orthopedics. Philadelphia:Saunders, 1990.
Resnick D, Niwayama I. Diagnosis of bone and jointdisorders. Philadelphia: Saunders, 1981.
Amir D, Mogle P, Weinberg H. Multiple epiphyseal dysplasiain one family, a further review of seven generations. J Bone Joint Surg (Br)1985; 67: 809-13.
Jacobs PA. Dysplasia epiphysialis multiplex. Clin Orthop1968; 58: 117-28.
Herring JA, Hotchkiss BL. Legg-Perthes disease versusmultiple epiphyseal dysplasia. J Pediatr Orthop 1987; 7:341-3.
Artikelinformatie
Citeer dit artikel als



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Reacties