
Samenvatting
Bij een meisje van 12,5 jaar, dat onder controle was vanwege diabetes mellitus type 1, werd een afbuigende lengtegroeicurve gezien met toename van het gewicht en moeilijk instelbare bloedsuikerwaarden. Er werd een van adrenocorticotroop hormoon (ACTH) onafhankelijke vorm van het syndroom van Cushing gediagnosticeerd. Tijdens de dexamethasonremmingstests was er een paradoxaal oplopen van de concentratie vrij cortisol in de urine. Hierdoor werd de diagnose ‘primaire gepigmenteerde nodulaire adrenocorticale ziekte’ (PPNAD) gesteld. De behandeling bestond uit bilaterale adrenalectomie en hormoonsubstitutie. PPNAD kan deel uitmaken van het zogenaamde carney-complex, een autosomaal dominant overervende aandoening met multipele neoplasieën. Screening van familieleden is derhalve aangewezen. Hierbij kan gebruik worden gemaakt van diagnostiek naar mutaties in het gen dat codeert voor de regulatoire subunit 1A van het proteïnekinase A (PRKAR1A).
Ned Tijdschr Geneeskd. 2006;150:2390-3
artikel
Inleiding
Zie ook de artikelen op bl. 2345, 2359 en 2365.
Het syndroom van Cushing komt niet vaak voor bij kinderen. In dit artikel beschrijven wij een casus met een bijzondere oorzaak, waarbij de toename van vrij cortisol in de urine tijdens de dexamethasonremmingstest de sleutel tot de diagnose gaf. Van belang is dat deze vorm van het cushingsyndroom kan passen binnen een familiaire aandoening met meerdere, ook endocriene, neoplasieën.
ziektegeschiedenis
Patiënt A, een meisje van 12,5 jaar, het eerste kind van gezonde ouders, werd op onze polikliniek gezien in verband met aanwijzingen voor het syndroom van Cushing. Zij was sinds 2,5 jaar bekend wegens diabetes mellitus type 1. Tijdens follow-up voor haar diabetes in het verwijzend ziekenhuis bleek de lengtegroei af te buigen en het gewicht toe te nemen. Ook waren de bloedsuikerwaarden moeilijk in te stellen en liep de concentratie geglycosyleerd hemoglobine (HbA1c) op. Daarbij ontstonden een vollemaansgezicht en een evidente vetafzetting op de romp. Omdat aan het syndroom van Cushing werd gedacht, werd de concentratie vrij cortisol in de 24-uursurine bepaald, welke waarde met 263 nmol/24 h licht verhoogd was (referentiewaarde: 30-250). Het dag-nachtritme van de serumcortisolwaarde was gestoord: de ochtendwaarde bedroeg 547 nmol/l en de waarde om 23:00 uur 669 nmol/l (normaal is de concentratie ’s ochtends het hoogst en om middernacht niet meetbaar laag). Bij de dexamethasonremmingstest met lage dosis (0,5 mg elke 6 h gedurende 48 h; bloedafname bij aanvang en na 48 h) en aansluitend met een hoge dosis (2 mg elke 6 h gedurende 48 h; bloedafname bij aanvang en na 48 h) bleek de serumcortisolspiegel niet te onderdrukken en toch werden zeer lage waarden van adrenocorticotroop hormoon (ACTH) gevonden. Tijdens deze tests nam de excretie van vrij cortisol in de 24-uursurine fors toe (tabel). Vanwege de zeer lage ACTH-waarden werd gedacht aan een adenoom of carcinoom van de bijnier. MRI van de bijnieren liet echter geen afwijkingen zien. Patiënte werd vervolgens naar onze polikliniek verwezen.
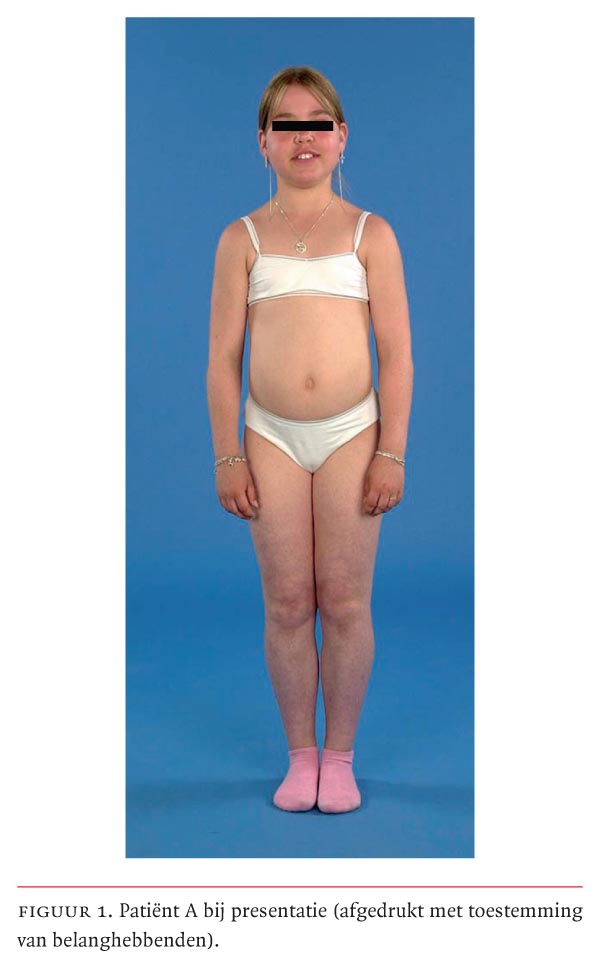
Bij lichamelijk onderzoek bedroeg de lichaamslengte van patiënte 141,1 cm (–2,5 SD), het gewicht 40,2 kg (+1,2 SD) en de bloeddruk 130/74 mmHg. Zij had een evidente cushingoïde habitus (figuur 1) en er was een toegenomen beharing op rug, armen en benen. Bij overig lichamelijk onderzoek werden geen bijzonderheden gevonden, met name bestonden er geen afwijkingen van de huid en de slijmvliezen. Patiënte was prepuberaal.
De biochemische uitslagen pasten bij een afwijking van de bijnier, maar bij beeldvormend onderzoek van de bijnieren werden geen bijzonderheden gevonden. Bijnierschorsscintigrafie met 131I-norcholesterol toonde symmetrische stapeling van norcholesterol in beide bijnieren. Er waren geen aanwijzingen voor ectopisch bijnierweefsel. Derhalve ging het om een autonome hyperactiviteit van beide bijnieren. Op basis van het paradoxaal oplopen van de excretie van vrij cortisol in 24-uursurine tijdens de dexamethasonremmingstest met lage en hoge doses dexamethason werd de waarschijnlijkheidsdiagnose ‘primaire gepigmenteerde nodulaire adrenocorticale ziekte’ (PPNAD) gesteld. Aangezien PPNAD in de meeste gevallen deel uitmaakt van het zogenaamde carney-complex, lieten wij nadere diagnostiek verrichten met echografie van de schildklier en echocardiografie. Beide onderzoeken brachten geen afwijkingen aan het licht.
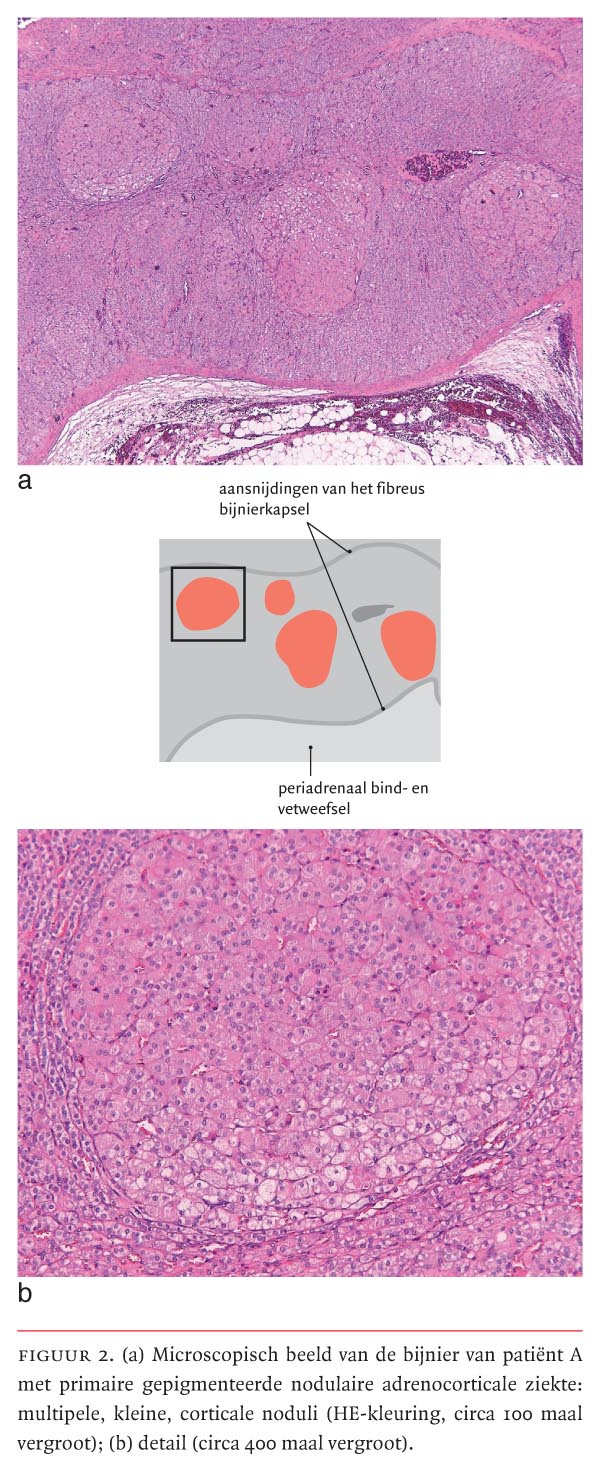
Wij verwezen patiënte naar onze kinderchirurgen voor bilaterale adrenalectomie. Bij pathologisch onderzoek van de bijnieren werd de diagnose ‘PPNAD’ bevestigd (figuur 2). Patiënte werd ingesteld op glucocorticoïd- en mineralocorticoïdsubstitutie en kon na enkele dagen het ziekenhuis in goede conditie verlaten. Na de operatie was er aanvankelijk een stilstand in de groei met verdere toename van het overgewicht tot 46 kg (+2 SD). Echter, 3 maanden na de operatie trad inhaalgroei op en nam het overgewicht af. In deze periode werd ook een geleidelijke verbetering van de bloedsuikerwaarden gezien. Tijdens de laatste controle op de polikliniek op de leeftijd van 14 jaar bedroeg de lengte van patiënte 148 cm (–2,5 SD), was haar gewicht 46 kg (+1 SD) en haar bloeddruk 110/65 mmHg. Zij was inmiddels in de puberteit.
Mutatieanalyse op een defect in het gen dat codeert voor de regulatoire subunit 1A van het proteïnekinase A (PRKAR1A), welk gen een rol speelt bij het carney-complex (zie verder), had bij patiënte en bij haar ouders een negatieve uitslag. Vooralsnog werden bij geen van de familieleden aanwijzingen voor het carney-complex gevonden.
beschouwing
PPNAD is een zeer zeldzame aandoening van beide bijnieren, die leidt tot een ACTH-onafhankelijke vorm van het syndroom van Cushing.1 Voor de kliniek, ontstaanswijze, diagnostiek en behandeling van het syndroom van Cushing bij kinderen verwijzen wij naar onze klinische les elders in dit nummer.2 PPNAD werd in 1984 voor de eerste maal beschreven.3 In de jaren daarvoor werd meestal gesproken over micronodulaire bijnierziekte.
PPNAD: diagnostiek, ontstaanswijze en behandeling
In de diagnostiek onderscheidt PPNAD zich van andere oorzaken van het syndroom van Cushing door het paradoxaal oplopen van de concentratie vrij cortisol in de urine tijdens de dexamethasonremmingstest.4 De oorzaak werd recent beschreven:5 in vitro blijkt toediening van dexamethason aan PPNAD-bijnierweefsel een toename van de glucocorticoïdsynthese te geven. Deze toename wordt vergezeld van een toegenomen expressie van glucocorticoïdreceptoren in de PPNAD-noduli ten opzichte van normale adrenocorticale cellen. De auteurs stellen dat de toename van de glucocorticoïdsecretie door dexamethason een direct en receptorspecifiek effect is. Het exacte mechanisme van receptoractivatie bij PPNAD is echter nog niet bekend. Beeldvormend onderzoek van de bijnieren is van beperkte waarde voor het diagnosticeren van PPNAD. De grootte van de bijnieren is in de meeste gevallen niet-afwijkend. Wel liet CT-onderzoek bij 48 van de patiënten met PPNAD en een niet-afwijkende grootte van de bijnieren een onregelmatige contour van deze bijnieren zien.4 Pathologisch onderzoek van de bijnieren toont multipele, kleine, macroscopisch en microscopisch donker gepigmenteerde corticale noduli, omgeven door internodulaire corticale atrofie. Het pigment is lipofuscine. De internodulaire atrofie is pathognomonisch voor PPNAD en weerspiegelt de autonome functie van deze noduli, met daardoor onderdrukte ACTH-spiegels.
De enige therapeutische optie voor patiënten met PPNAD is bilaterale adrenalectomie.
PPNAD en het carney-complex
PPNAD kan geïsoleerd voorkomen, maar bij 95 van de patiënten maakt het deel uit van het carney-complex.6 Dit is een syndroom van multipele neoplasieën, waarbij cardiale, endocriene, cutane, gonadale en neurale tumoren kunnen voorkomen. Daarnaast kan een scala aan gepigmenteerde afwijkingen van huid en mucosa bestaan. De overerving is autosomaal dominant, derhalve worden de meeste casussen binnen families beschreven. In dat kader is het belangrijk familieleden te screenen op aspecten van het carney-complex.
Kliniek van het carney-complex
De klinische manifestatie kan bij patiënten van dezelfde familie verschillen en hangt mede samen met de leeftijd bij presentatie. De huidafwijkingen kunnen bij de geboorte reeds aanwezig zijn, maar ontwikkelen zich meestal tijdens de kinderjaren. Op de zuigelingenleeftijd komen cardiale en cutane myxomen en PPNAD het meest voor. Schildkliernoduli ontstaan meestal in het eerste decennium. PPNAD komt van de endocriene tumoren het frequentst voor, en wordt bij ongeveer een kwart van de patiënten beschreven.6
Genetica
Recent onderzoek naar de genetische achtergrond van PPNAD en het carney-complex toonde bij 82 van de patiënten een defect in PRKAR1A.7 Dit gen, gelokaliseerd op chromosoom 17q22-24, werd in 2000 geïdentificeerd en lijkt te functioneren als een tumorsuppressorgen.8 Afname van de functie van PRKAR1A leidt tot toename van de proteïnekinase A-activiteit, met als gevolg tumorgenese. Inactiverende mutaties in het PRKAR1A-gen worden bij ongeveer 40 van de patiënten met het carney-complex beschreven.9 Bij volwassen patiënten met het carney-complex die zich presenteren met het syndroom van Cushing door PPNAD, ligt dit waarschijnlijk rond de 80.7 Wanneer PPNAD geïsoleerd voorkomt, bedraagt de mutatiefrequentie 65.10 Mogelijk is ook een ander gen, gelegen op chromosoom 2p16, betrokken bij de pathogenese van het carney-complex.11
conclusie
PPNAD is een zeldzame oorzaak van een ACTH-onafhankelijke vorm van het syndroom van Cushing bij kinderen. In de diagnostiek van PPNAD is het paradoxaal oplopen van de concentratie vrij cortisol in de urine tijdens de dexamethasonremmingstest zeer specifiek. PPNAD is bij de meeste patiënten onderdeel van het carney-complex, een autosomaal dominant overervend multipele-neoplasiesyndroom. De klinische manifestatie van de ziekte kan wisselen, ook binnen dezelfde familie. Screening van familieleden op aspecten van het carney-complex is derhalve aangewezen. Genetisch onderzoek naar mutaties in het PRKAR1A-gen kan daarbij van waarde zijn.
Belangenconflict: geen gemeld. Financiële ondersteuning: geen gemeld.
Literatuur
Stratakis CA, Kirschner LS. Clinical and genetic analysis of primary bilateral adrenal diseases (micro- and macronodular disease) leading to Cushing syndrome. Horm Metab Res. 1998;30:456-63.
Bocca G, Voorhoeve PG, Delemarre-van de Waal HA. Het syndroom van Cushing bij kinderen. Ned Tijdschr Geneeskd. 2006;150:2345-9.
Shenoy BV, Carpenter PC, Carney JA. Bilateral primary pigmented nodular adrenocortical disease. Rare cause of the Cushing syndrome. Am J Surg Pathol. 1984;8:335-44.
Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131:585-91.
Bourdeau I, Lacroix A, Schürch W, Caron P, Antakly T, Stratakis CA. Primary pigmented nodular adrenocortical disease: paradoxical responses of cortisol secretion to dexamethasone occur in vitro and are associated with increased expression of the glucocorticoid receptor. J Clin Endocrinol Metab. 2003;88:3931-7.
Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041-6.
Groussin L, Kirschner LS, Vincent-Dejean C, Perlemoine K, Jullian E, Delemer B, et al. Molecular analysis of the cyclic AMP-dependent protein kinase A (PKA) regulatory subunit 1A (PRKAR1A) gene in patients with Carney complex and primary pigmented nodular adrenocortical disease (PPNAD) reveals novel mutations and clues for pathophysiology: augmented PKA signaling is associated with adrenal tumorigenesis in PPNAD. Am J Hum Genet. 2002;71:1433-42.
Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS, et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89-92.
Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the Carney complex. Hum Mol Genet. 2000;9:3037-46.
Groussin L, Cazabat L, René-Corail F, Jullian E, Bertherat J. Adrenal pathophysiology: lessons from the Carney complex. Horm Res. 2005;64:132-9.
Matyakhina L, Pack S, Kirschner LS, Pak E, Mannan P, Jaikumar J, et al. Chromosome 2 (2p16) abnormalities in Carney complex tumours. J Med Genet. 2003;40:268-77.
Artikelinformatie
Citeer dit artikel als




{kind=link}
{kind=link}
{kind=link}
Reacties